D22
Large dynamic range small-angle diffractometer
Instructions for the use of CRYSON
Below you will find short instructions how to use CRYSON by Dmitri Svergun. Instructions for CRYSOL are here.
The information below corresponds to the file /hosts/lass2/d1/lss/svergun/Crysol/cryson.ins.
At the ILL, use the command cryson.
C R Y S O N
This is a short information on how to use beta-release of Cryson,
which is a version of CRYSOL for neutron scattering.
IBM-PC version:
Hardware requirements: IBM-PC compatible (486 and higher)
with 2 Mb RAM and VGA or SVGA graphic card.
Operating systems:
DOS/Win3.1/Win95/WinNT.
The program works in a full-screen DOS window.
Under Win3.1 one should call a full-screen DOS prompt.
Under Win95, one should invoke a Real DOS mode.
Under WinNT, one should reboot in DOS mode or
switch off the EMS usage in the properties of the DOS window.
The programs CRYSON.EXE requires the same DOS-extender
as Crysol, namely, DOSXMSF.EXE (Phar Lap), and also
the font file COURB.FON to be in the current directory
or in the root directory C:\.
SGI version was compiled under IRIX 6.4, Fortran compiler 7.4
For the general information see file Crysol.ins in the Crysol
distribution file. The differences between Crysol and Cryson are:
(i) instead of the solvent density in e/A**3 required by
Crysol, Cryson prompts for the D2O content in the solvent.
The solvent density SD is then evaluated as
SD = ROH2O*(1.- Y) + ROD2O*Y
where Y is the D2O concentration (ranges from 0 to 1)
ROH2O = 0.562 is the H2O scattering length density (10**10 cm**-2)
ROD2O = 6.404 is the D2O scattering length density (10**10 cm**-2)
The user may change the solvent density value manually.
(ii) if Y is not zero, Cryson prompts for the fraction of nonexchanged NH
peptide DNEXCH. It is assumed that all exchangeable hydrogens (which
belong to NH, NH2, NH3, OH and SH groups) are
replaced by deuteriums in proportion Y, except for the main-chain NH
groups, for which the probability is Y*(1-DNEXCH). Default value
DNEXCH = 0.1 is generally accepted. The scattering length density B of
any atom/atomic group at the D2O concentration Y is evaluated as
B(Y) = B(0) + EXCH * Y * ( BD - BH ) * Afac
where B(0) is the scattering length in H2O,
BH = -0.3742 is the scattering length of hydroden (10-12 cm),
BD = 0.6671 is the scattering length of deuterium (10-12 cm),
EXCH is the number of exchangeable hydrogens in the group,
Afac = 1-DNEXCH for the main chain NH groups, otherwise 1.
The scattering lengths and the number of exchangeable hydrogens are taken as below:
Scattering lengths at 0%D2O (in 10-12 cm)
C | CH | CH2 | CH3 | N | NH | NH2 | NH3 | ||||
.6651 | .2909 | .083 | .458 | .940 | .5658 | .1916 | .1826 | ||||
O | OH | S | SH | P | FE | CU | CA | MG | MN | ZN | N(Histidine) |
.5804, | .2062, | .2847, | .090, | .5170, | .95 | .76 | .47 | .52 | .39 | .57 | .7529 |
Number of exchangeable protons
C | CH | CH2 | CH3 | N | NH | NH2 | NH3 | O | OH |
0. | 0. | 0. | 0. | 0. | 1. | 2. | 3. | 0. | 1. |
S | SH | P | FE | CU | CA | MG | MN | ZN | N(Histidine) |
0. | 1. | 0. | 0. | 0. | 0. | 0. | 0. | 0. | 0.5 |
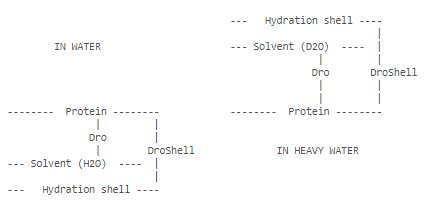
- by default, Cryson searches for the contrast in the hydration shell in the range 0 < DroShell < 0.2*ABS(SD) [10**10 cm**-2]. For proteins in H2O and D2O, positive DroShell means more dense solvent in the shell, as seen from the figure below. One can, however, also imagine the situation when the positive DroShell would mean less dense solvent in the shell. The user can perform the search both in positive and negative ranges of DroShell, if necessary.
The units for the contrasts and the scattering length densities are always 10**10 cm-2, the intensities in the *.int file are in cm**2.
(iii) CRYSON also fits a flat background. The value to be subtracted/added is searched for in units of an average of the ten last experimental points.
(iv) CRYSON smears the theoretical curves using the resolution function introduced by J.Skov Pedersen et al. (1990), J.Appl. Cryst., 23, 321 Several subroutines for data smearing are provided by J.Skov Pedersen and modifies for the use in CRYSON. The program prompts for a resolution file which should have the following format (the numbers describe a setting at RISOE SANS instrument):
0.8 , Effective collimation slit diameter in cm
0.35 , Effective sample diameter in cm
300. , Collimation distance in cm
105. , Sample-detector distance in cm
3. , Lambda in Angstroems
0.18 , Delta(Lambda)/Lambda
1.1 , Pixel size in cm
0.0000 , Averaging error (accounted for in Pixel size)
If the file is corrupted or does not exist, no smearing is performed.
An example of the resolution file is provided (file ill.res).
- Back to the D22 documentation
- Back to the LSS home page