Exploring the protein folding chaperones at the heart of our cells
Insulin is a hormone that is rapidly secreted into the blood, following a meal, in order to control blood glucose levels. These pulses of insulin secretion enable the body to take up and store sugar and quickly return blood glucose to normal levels. Diabetes, which affects 1 in 10 people in Europe, occurs when this process goes wrong.
The endoplasmic reticulum (ER), a protein-producing compartment contained within almost every animal cell, is responsible for the synthesis of insulin and up to one third of all encoded proteins. One of the major agents responsible for helping the folding of nascent proteins into their correct and functional 3D shape is BiP, an example of a protein chaperone.
It is important for a cell to have enough BiP chaperone proteins available to deal with its requirements for the production and folding of proteins destined for secretion. Having a folding imbalance, between the number of proteins which require folding and the amount of available protein chaperones, can result in the accumulation of unfolded or misfolded proteins. This can put considerable stress on a cell. If enough cells experience excessive and prolonged stress this can result in tissue death and serious disease.
As a testament to the importance of maintaining a balance between chaperones and unfolded proteins, animal cells have evolved a number of ways to modulate the amount and activity of BiP within the ER.
One method is through direct modification of BiP using a molecule of adenosine monophosphate (AMP). In order to match the protein folding demands of the ER, an enzyme called FICD is able to rapidly add (AMPylate) or remove (deAMPylate) this AMP molecule — thereby inactivating or reactivating the chaperone, respectively.
The precise structure and mechanism of how BiP interacts with and is modified/de-modified by FICD has until now remained obscure. For the first time, scientists have captured a snapshot of the evolutionarily conserved complex formed between BiP and FICD, providing detailed insights into the both the enzymatic mechanisms of FICD’s two opposing activities and the behaviour of the BiP-FICD complex in solution.
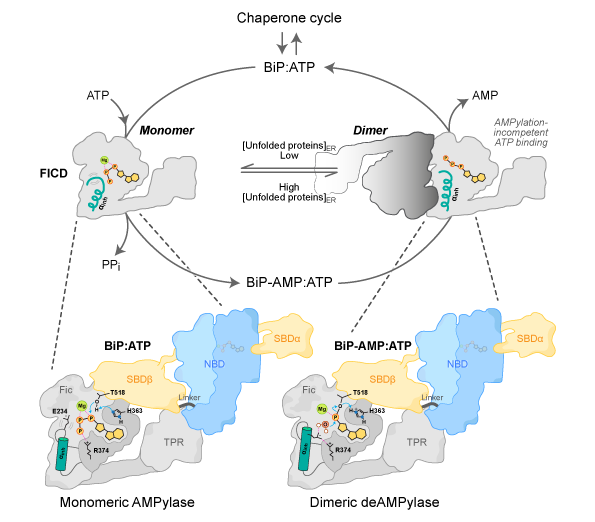
It is speculated that the FICD monomer-dimer equilibrium is adjusted in response to changing levels of unfolded proteins within the endoplasmic reticulum by processes which may include a direct response to changes in the endoplasmic reticulum energy status (ATP/ADP ratio). Adapted, with permission, from Perera et al., 2021.
The Institut Laue-Langevin (ILL) offers some of the world’s most advanced analysis techniques employing neutrons. By illuminating samples with an intense beam of neutrons, scientists can interpret the subsequent scattering pattern and infer their organisation at the nanometer scale.
Small Angle Neutron Scattering (SANS) is perfectly suited to exploring biological matter under close to physiological conditions in solution. This contrasts with other techniques which probe the structure of proteins, such as crystallography (which requires proteins to pack into a rigid crystalline state) or Cryo-EM (in which the sample must be frozen in a vitreous state).
A team of scientists from the University of Cambridge and the ILL approached this research with the goal of solving the structure of a complex of BiP and FICD, first using crystallography and then in solution with SANS. Using the D11 instrument at ILL, they validated and extended their crystallographic findings — confirming the anticipated size, shape and arrangement of the protein complex and its constituents. In addition, SANS also provided indications of structural flexibility within the protein complex, information which was inaccessible to the static pictures provided by crystallography alone.
A process called deuteration — replacement of hydrogen atoms with a heavier deuterium isotope — formed an essential part of the protein sample preparation and was carried out within the ILL’s Deuteration Laboratory. This enabled the formation of protein complexes in which only one of the two proteins in the complex was (isotopically) modified. The aqueous solutions were further prepared with varying ratios of normal and deuterated (heavy) water.
Substitution of hydrogen with deuterium in the solvent modulates the “contrast” of the protein components with the surrounding solution. In this study, scientists used the phenomenon of contrast matching to cause individual elements of the protein complex to appear ‘invisible’ to neutrons, thereby revealing structural details of individual proteins within the context of the whole complex.
These insights contribute to expanding the scientific community’s understanding of how protein folding activity in the ER is modulated by FICD. In the future, this deeper understanding may help us to better comprehend, and possibly treat, human diseases associated with instances of improper protein folding within the ER.
Structures of a deAMPylation complex rationalise the switch between antagonistic catalytic activities of FICD (2021)
doi: 10.1038/s41467-021-25076-7
ILL Instrument: ILL’s Small-Angle Neutron Scattering instrument D11 was used in this experiment.
Contact:Luke Perrera (University of Cambridge)
ILL contact: Sylvain Prevost